Load genome-wide binding data for gene expression (RNA polymerase occupancy)
Source:R/load_data.R
load_data_genes.RdReads RNA Polymerase DamID binding profiles either from bedGraph files or directly from a list of GRanges objects. Calculates binding occupancy summarised over genes.
Usage
load_data_genes(
binding_profiles_path = NULL,
binding_profiles = NULL,
drop_samples = NULL,
norm_method = c("none", "loess", "quantile", "rpm"),
pre_scale = FALSE,
quantile_norm = NULL,
organism = "drosophila melanogaster",
calculate_occupancy_pvals = TRUE,
return_per_replicate_fdr = FALSE,
occupancy_plot_diagnostics = interactive(),
null_model_iterations = 1e+05,
ensdb_genes = NULL,
BPPARAM = BiocParallel::bpparam(),
plot_diagnostics = interactive()
)Arguments
- binding_profiles_path
Character vector of directories or file globs containing log2 ratio binding tracks in bedGraph format. Wildcards ('*') supported.
- binding_profiles
A list of GRanges objects (one per sample) or a single GRanges object containing multiple sample metadata columns.
- drop_samples
A character vector of sample names or patterns to remove. Matching samples are removed from the analysis before normalisation and occupancy calculation. This can be useful for excluding samples that fail initial quality checks. Default: `NULL` (no samples are dropped).
- norm_method
Character. Determines how raw genome-wide signal is normalised prior to establishing peak specific occupancy. Options are:
"none"(default): Use original data. Recommended if loading CATaDa count data pre-processed viadamidseq_pipeline --catada(which is natively RPM-normalised)."loess": Uses the `cyclicloess` method from `limma`."quantile": Quantile normalisation. Forces identical target distributions."rpm": Reads Per Million scaling. A simple global scaling for raw un-normalised count data. Use with caution: do not use for log2 ratio data, and avoid if CATaDa data has already been RPM normalised (the default behaviour with `damidseq_pipeline`)
- pre_scale
Logical. If TRUE, samples will be scaled without centering via base R `scale(center=FALSE, scale=TRUE)`, prior to normalisation being applied. Default is FALSE.
- quantile_norm
Deprecated. Please use `norm_method = "quantile"` instead.
- organism
Organism string (lower case) to obtain genome annotation from (if not providing a custom `ensdb_genes` object) Defautls to "drosophila melanogaster".
- calculate_occupancy_pvals
Calculate occupancy p-values as a proxy for gene expression status (see details). Not used for differential expression analysis, but used when present for downstream analysis and plotting. (default: TRUE)
- return_per_replicate_fdr
Legacy option of returning BH-adjusted RNA Polymerase occupancy FDR values per replicate. As of v0.99.12, unadjusted p-values are returned by defualt; these are then aggregated at the condition level during `differential_binding()` and the aggregate p-values adjusted to gain statistical power. This option exists for legacy or unsual end-user applications. Use with caution. (default: FALSE)
- occupancy_plot_diagnostics
Logical. If `TRUE` (default in interactive sessions), diagnostic plots for the gene expression null model will be displayed.
- null_model_iterations
Number of iterations to use to determine null model for FDR (default: 100000)
- ensdb_genes
GRanges object: gene annotation. Automatically obtained from `organism` if NULL.
- BPPARAM
BiocParallel function (defaults to BiocParallel::bpparam())
- plot_diagnostics
Logical. If `TRUE` (the default in interactive sessions), diagnostic plots (PCA and correlation heatmap) will be generated and displayed for both the raw binding data and the summarised occupancy data.
Value
List with elements:
- binding_profiles_data
GRanges: Merged binding profiles, with genomic coordinates and sample metadata columns.
- occupancy
data.frame: Occupancy values summarised over genes.
- test_category
Character scalar; will be "expressed".
- metadata
List: Data provenance and analysis metadata.
Details
One of `binding_profiles_path` or `binding_profiles` must be provided.
When supplying GRanges lists for `binding_profiles`, each GRanges should contain exactly one numeric metadata column representing the binding signal, and `binding_profiles` must be a named list, with element names used as sample names. Alternatively, a single GRanges object already containing multiple sample columns may be provided.
Examples
# Create a mock GRanges object for gene annotations
# This object avoids network access
# and includes a very long gene to ensure overlaps with sample data.
mock_genes_gr <- GenomicRanges::GRanges(
seqnames = S4Vectors::Rle("2L", 7),
ranges = IRanges::IRanges(
start = c(1000, 2000, 3000, 5000, 6000, 7000, 8000),
end = c(1500, 2500, 3500, 5500, 6500, 7500, 20000000)
),
strand = S4Vectors::Rle(GenomicRanges::strand(c("+", "-", "+", "+", "-", "-", "+"))),
gene_id = c("FBgn001", "FBgn002", "FBgn003", "FBgn004", "FBgn005", "FBgn006", "FBgn007"),
gene_name = c("geneA", "geneB", "geneC", "geneD", "geneE", "geneF", "LargeTestGene")
)
# Get path to sample data files included with the package
data_dir <- system.file("extdata", package = "damidBind")
# Run loading function using sample files and mock gene annotations
# This calculates occupancy over genes instead of peaks.
loaded_data_genes <- load_data_genes(
binding_profiles_path = data_dir,
ensdb_genes = mock_genes_gr,
norm_method = "none",
calculate_occupancy_pvals = FALSE
)
#> Locating binding profile files
#> Building binding profile dataframe from input files ...
#> - Loaded: Bsh_Dam_L4_r1-ext300-vs-Dam.kde-norm
#> - Loaded: Bsh_Dam_L4_r2-ext300-vs-Dam.kde-norm
#> - Loaded: Bsh_Dam_L5_r1-ext300-vs-Dam.kde-norm
#> - Loaded: Bsh_Dam_L5_r2-ext300-vs-Dam.kde-norm
#> Calculating average occupancy for 7 regions...
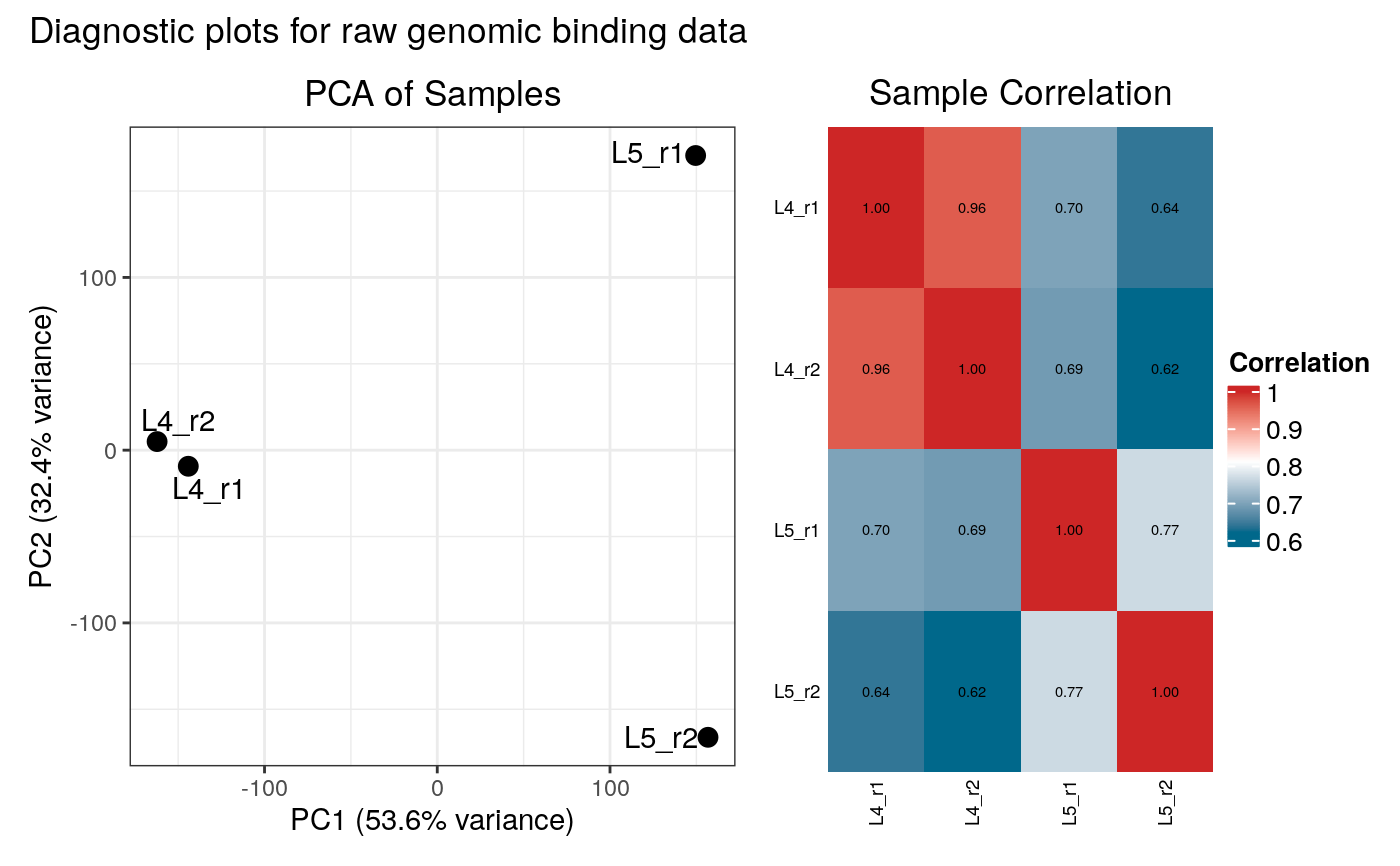
#> Generating diagnostic plots...
#> 3 rows with zero variance were filtered.
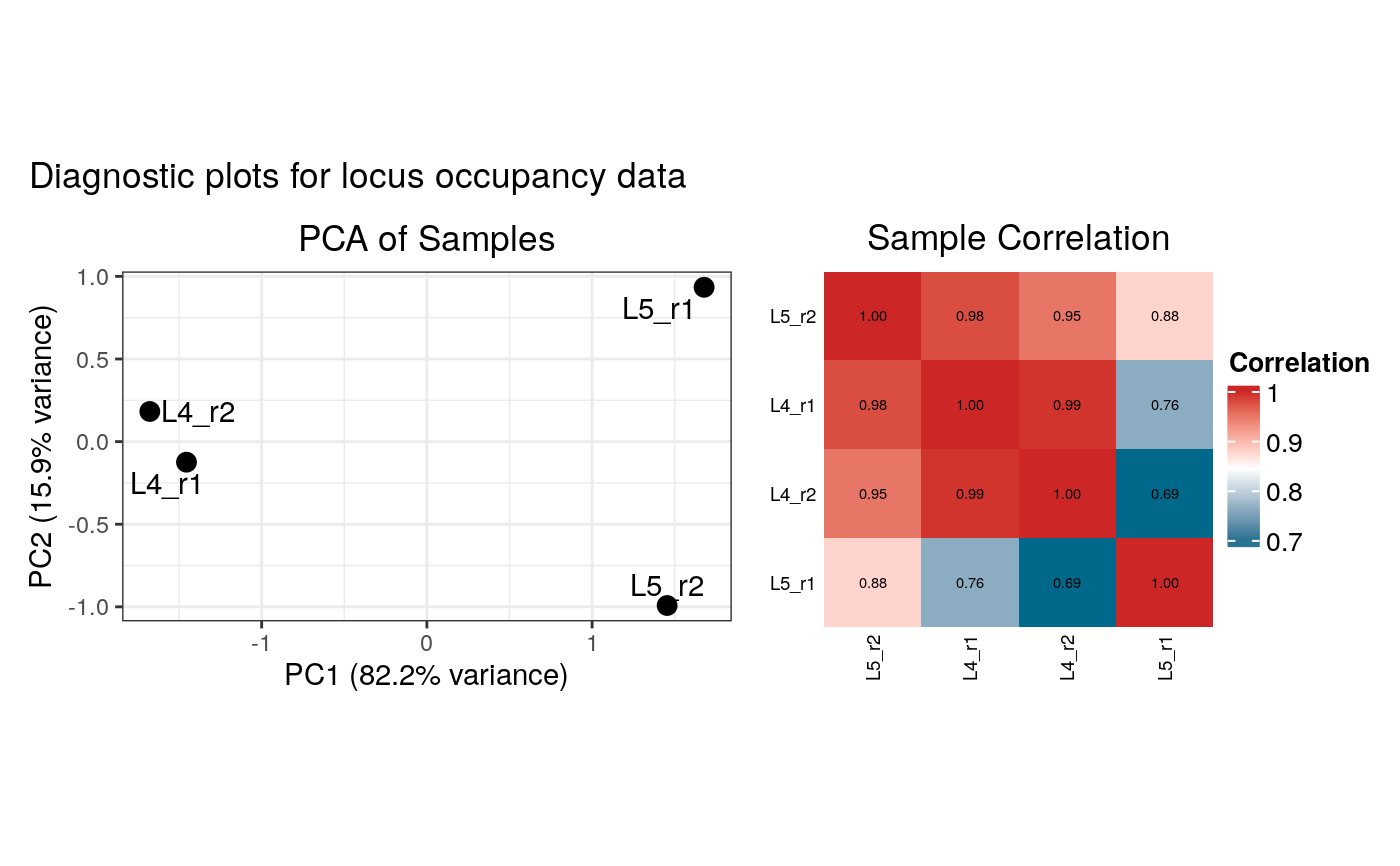 #> 3989 rows with zero variance were filtered.
#> 3989 rows with zero variance were filtered.
 # View the head of the occupancy table
head(loaded_data_genes$occupancy)
#> gene_id gene_name name nfrags
#> 2L:1000-1500 FBgn001 geneA 2L:1000-1500 4
#> 2L:2000-2500 FBgn002 geneB 2L:2000-2500 4
#> 2L:3000-3500 FBgn003 geneC 2L:3000-3500 4
#> 2L:5000-5500 FBgn004 geneD 2L:5000-5500 4
#> 2L:6000-6500 FBgn005 geneE 2L:6000-6500 2
#> 2L:7000-7500 FBgn006 geneF 2L:7000-7500 1
#> Bsh_Dam_L4_r1-ext300-vs-Dam.kde-norm
#> 2L:1000-1500 0.000000
#> 2L:2000-2500 0.000000
#> 2L:3000-3500 0.000000
#> 2L:5000-5500 1.465469
#> 2L:6000-6500 1.515389
#> 2L:7000-7500 -0.130000
#> Bsh_Dam_L4_r2-ext300-vs-Dam.kde-norm
#> 2L:1000-1500 0.000000
#> 2L:2000-2500 0.000000
#> 2L:3000-3500 0.000000
#> 2L:5000-5500 1.444371
#> 2L:6000-6500 1.791916
#> 2L:7000-7500 -0.110000
#> Bsh_Dam_L5_r1-ext300-vs-Dam.kde-norm
#> 2L:1000-1500 0.0000000
#> 2L:2000-2500 0.0000000
#> 2L:3000-3500 0.0000000
#> 2L:5000-5500 1.2137325
#> 2L:6000-6500 0.2373453
#> 2L:7000-7500 -0.0300000
#> Bsh_Dam_L5_r2-ext300-vs-Dam.kde-norm
#> 2L:1000-1500 0.0000000
#> 2L:2000-2500 0.0000000
#> 2L:3000-3500 0.0000000
#> 2L:5000-5500 0.8943313
#> 2L:6000-6500 0.6489222
#> 2L:7000-7500 -0.0900000
# View the head of the occupancy table
head(loaded_data_genes$occupancy)
#> gene_id gene_name name nfrags
#> 2L:1000-1500 FBgn001 geneA 2L:1000-1500 4
#> 2L:2000-2500 FBgn002 geneB 2L:2000-2500 4
#> 2L:3000-3500 FBgn003 geneC 2L:3000-3500 4
#> 2L:5000-5500 FBgn004 geneD 2L:5000-5500 4
#> 2L:6000-6500 FBgn005 geneE 2L:6000-6500 2
#> 2L:7000-7500 FBgn006 geneF 2L:7000-7500 1
#> Bsh_Dam_L4_r1-ext300-vs-Dam.kde-norm
#> 2L:1000-1500 0.000000
#> 2L:2000-2500 0.000000
#> 2L:3000-3500 0.000000
#> 2L:5000-5500 1.465469
#> 2L:6000-6500 1.515389
#> 2L:7000-7500 -0.130000
#> Bsh_Dam_L4_r2-ext300-vs-Dam.kde-norm
#> 2L:1000-1500 0.000000
#> 2L:2000-2500 0.000000
#> 2L:3000-3500 0.000000
#> 2L:5000-5500 1.444371
#> 2L:6000-6500 1.791916
#> 2L:7000-7500 -0.110000
#> Bsh_Dam_L5_r1-ext300-vs-Dam.kde-norm
#> 2L:1000-1500 0.0000000
#> 2L:2000-2500 0.0000000
#> 2L:3000-3500 0.0000000
#> 2L:5000-5500 1.2137325
#> 2L:6000-6500 0.2373453
#> 2L:7000-7500 -0.0300000
#> Bsh_Dam_L5_r2-ext300-vs-Dam.kde-norm
#> 2L:1000-1500 0.0000000
#> 2L:2000-2500 0.0000000
#> 2L:3000-3500 0.0000000
#> 2L:5000-5500 0.8943313
#> 2L:6000-6500 0.6489222
#> 2L:7000-7500 -0.0900000